
Molecules, which are made up of atoms, are complex quantum systems that play a crucial role in various fields, such as drug development and material design. The computer simulation of molecules has long been a challenging task due to the intricate interactions of electrons within them. However, a recent breakthrough by researchers from the Berlin Institute for the Foundations of Learning and Data (BIFOLD) at TU Berlin and Google DeepMind has paved the way for highly accurate simulations of molecular dynamics on long time-scales.
Traditional methods for computing electron interactions in molecules rely on solving the Schrödinger equation, which describes the energy levels that a quantum system can assume. This process is extremely difficult and time-consuming, especially for molecules with a large number of atoms. Running molecular dynamics simulations further exacerbates this challenge, as the Schrödinger equation needs to be solved numerous times, consuming substantial computational resources.
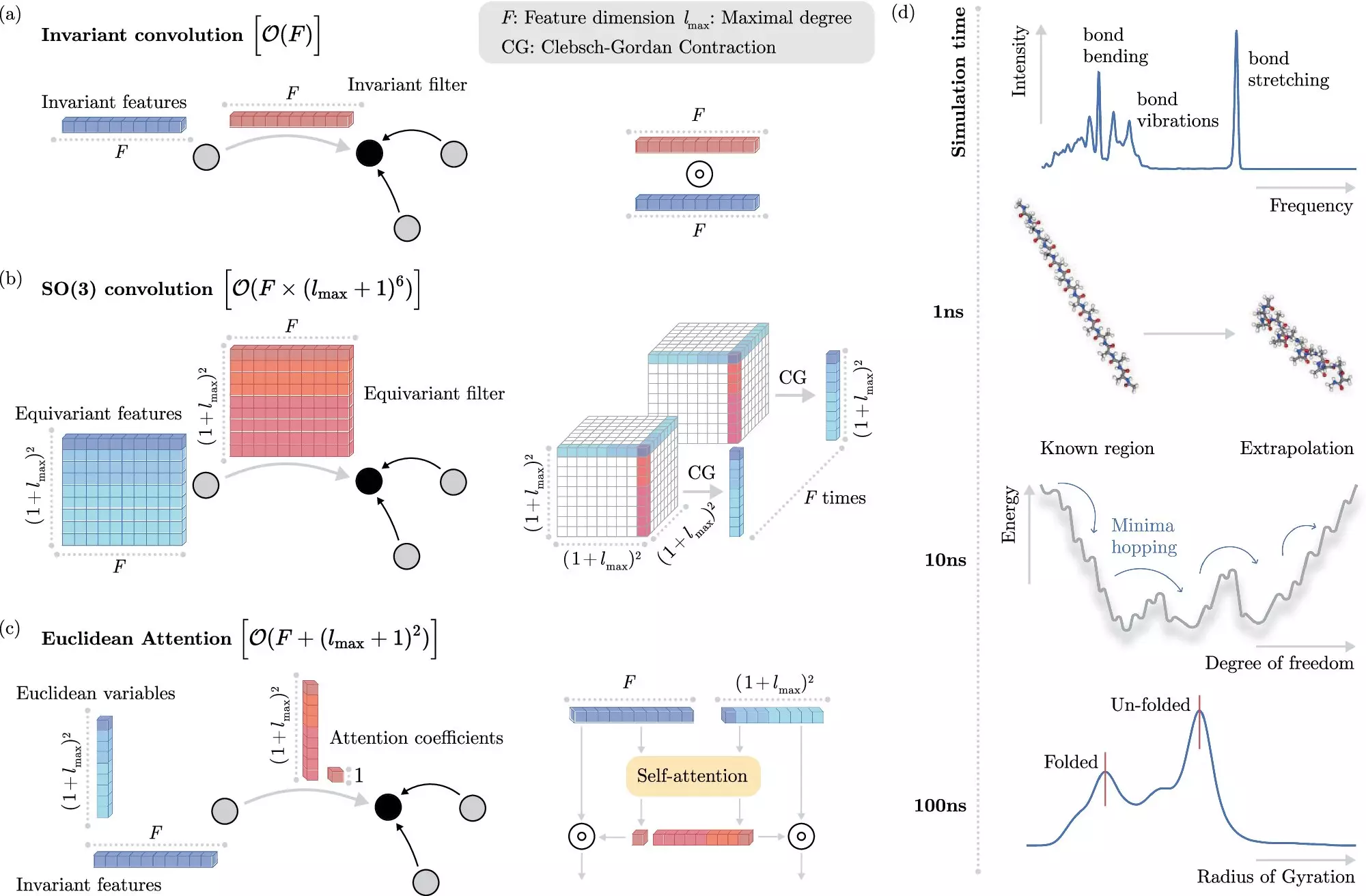
In recent years, machine learning (ML) methods have emerged as a promising solution to this problem. Instead of explicitly solving the Schrödinger equation, ML algorithms can learn to predict electronic interactions at the atomistic level, significantly reducing computational costs. However, incorporating physical invariances into ML models has proven to be computationally expensive, limiting the speed of molecular dynamics simulations.
To address this limitation, researchers at BIFOLD have developed a novel learning algorithm that separates invariances from other information about a chemical system. This innovative approach allows the ML model to focus on essential physical information, thus streamlining complex operations and reducing overall computational costs. As a result, simulations that previously took months or years to complete can now be executed within days on a single computer node.
The efficiency gained from this new algorithm opens up a wide array of possibilities in the field of molecular dynamics simulations. Researchers can now explore long-time scale simulations to gain deeper insights into the structure and functioning of atomistic systems. Moreover, the accurate simulation of molecular interactions could revolutionize drug development, saving time, money, and resources while promoting environmental sustainability.
Looking ahead, the combination of advanced machine learning techniques with physical principles is expected to drive further advancements in computational chemistry. The ability to accurately simulate complex molecular systems will be crucial for developing new drugs and understanding fundamental processes in nature. The next generation of algorithms will need to tackle even larger and more complex system sizes, requiring a precise description of intricate long-range physical interactions.
The groundbreaking work by researchers at BIFOLD and Google DeepMind represents a significant leap forward in the field of molecular dynamics simulations. By harnessing the power of machine learning, scientists are now able to explore the dynamics of molecules in ways that were previously unattainable. This innovative approach not only accelerates research efforts but also holds immense potential for shaping the future of drug discovery and material design.
Leave a Reply
You must be logged in to post a comment.